设计具有目标吸附性能催化活性位点的催化材料是异质催化研究的核心科学问题,直接影响能源转化效率、反应选择性与材料可控合成等关键技术的突破。尽管近年来以高通量密度泛函理论(DFT)计算和AI机器学习为基础的正向设计方法已取得显著进展,但在结构复杂、动态演化显著的催化体系中,如何实现从性能反推结构的逆向设计仍面临三大挑战:结构空间维度爆炸、模型可解释性不足以及生成能力受限。
图论结构化学作为一种将材料微观结构映射为数学图模型的方法,近年来在材料基因组和催化活性探索等领域展现出卓越的表征能力。潘锋教授团队长期致力于图论结构化学方法的拓展与应用,并在该领域取得了一系列创新性工作,实现材料结构表示(Sci China Chem, 2019, DOI: 10.1007/s11426-019-9502-5),开发低维材料设计方法(National Science Review, 2022, DOI: 10.1093/nsr/nwac028),创制新型固态电解质(J. Am. Chem. Soc. 2024, 146, 27, 18535–18543),实现催化反应路径搜索(CCS Chemistry 2024, 7, 1-14 DOI:10.31635/ccschem.024.202404955),发现催化活性相搜索(Nat Commun 16, 2542 (2025). DOI:10.1038/s41467-025-57824-4)。
近日,潘锋团队联合北京数学科学与应用研究院(由国际数学家丘成桐先生创立)李京艳助理研究员及厦门大学郑世胜特任副研究员等,在图论结构化学的基础上创新融合代数拓扑数据分析与深度生成AI建模技术,成功构建了一种具有物理可解释性的催化活性位点逆向设计框架。相关研究成果以“Inverse design of catalytic active sites via interpretable topology-based deep generative models”为题,发表于Nature 旗下知名子刊《npj Computational Materials》(2025, 11:147)。
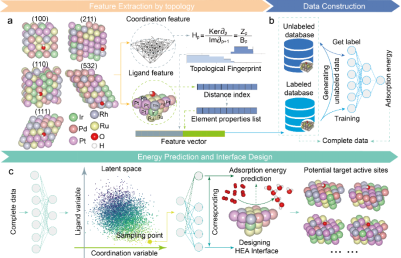
催化活性位点逆向设计的PGH-VAEs框架及建模流程
GLMY同调(persistent GLMY homology, PGH)由国际著名数学家丘成桐先生团队提出,是一种能够处理非对称、有向图结构的代数拓扑方法,特别适用于复杂结构体系的几何—拓扑建模。复杂的催化活性位点逆向设计框架是基于图论结构化学对原子级结构的抽象建模,以持续GLMY同调为核心拓扑工具,从几何结构中提取拓扑不变量,能够刻画活性位点的三维连通性与空穴等关键结构信息。研究团队以IrPdPtRhRu高熵合金为研究对象,创新性开发了融合PGH拓扑指纹与元素配体信息的双通道结构表征方法,分别编码原子配位与远场调控效应,结合变分自编码器(VAE)构建了结构—性能关联生成模型。在训练数据有限(仅约1100条DFT样本)的条件下,通过引入半监督学习机制和梯度提升回归器(GBRT),模型对*OH吸附能的预测平均绝对误差达到0.045 eV,展现出极高的预测精度。
该研究首次揭示PGH拓扑特征(Betti数)与吸附性能之间存在显著线性相关性,特别是原子连通性(0维)与结构空穴(2维)对性能调控起决定性作用。这一发现不仅为数字潜空间提供了物理解释依据,更显著提升了结构采样的针对性。在结构生成过程中,模型成功识别出以Pt/Pd为桥位、Ru为远场调控原子的最优构型,并进一步预测(111)和(211)晶面的最优组分比例(分别为0.7:0.2:0.1与3:3:4),为催化剂合成实验提供了定量指导。
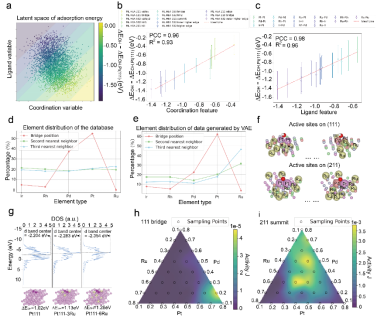
PGH拓扑特征与吸附性能的相关性分析与代表性生成结构示意
该研究展示了图论结构化学+代数拓扑+深度生成AI模型三者协同研究的潜力,为异质催化领域提供了一种面向结构—性能建模与从性能到结构逆向设计的新范式。因此,该框架不仅适用于高熵合金催化剂体系,还可扩展至电还原、氧化反应等多种催化场景的结构建模与活性探索。
潘锋教授、李京艳助理研究员与郑世胜副研究员为本研究的共同通讯作者,北京大学深圳研究生院新材料学院博士研究生王炳胥为第一作者。该工作得到了国家自然科学基金、电动汽车动力电池与材料国际联合研究中心、广东省新能源材料设计与计算重点实验室、深圳市新能源材料基因组制备和检测重点实验室的支持。


