机器学习在各领域的广泛应用促生其在材料领域的应用,它提供了一种新型的工具,即能从高维数据中发现数据间的规律,有助于减少计算量从而加速对新材料的探索。特征提取(特征工程)是机器学习的关键组成部分,选择合适的形式来表达将直接影响最终模型的效果。在材料科学领域必须要捕获所有的相关信息,从而达到区分不同原子和不同晶体环境的作用。因此在材料领域中,特征可以是简单的确定原子序数,可能涉及复杂的转换,比如径向分布函数(RDFs)的扩展,也可能是聚合统计数据(例如求平均值、求最大值等)。
在数学中,拓扑可以用来处理空间中不同组件的连通性,并表征空间中独立的实体,环和高维拓扑面。拓扑提供了最高级别的抽象因此可以作为一种处理高维结构数据的有效工具。其中,拓扑数学的持续同调(persistent homology)通过将多尺度几何信息嵌入拓扑不变量从而将几何分析和拓扑分析连接起来,它是一种在变化的尺度中分析拓扑结构的一种重要的方法。基本思想是随着原子尺度的变化记录结构的拓扑不变量(例如β0,β1和 β2),从而得到拓扑指纹(如图1所示)。


图1 类苯环结构的拓扑指纹示意图及碳硼烷结构的预测
在此过程中,利用持续时间较长的拓扑特征来表征系统的内在特性,通过预先设定的阈值进行过滤。在三维空间中,独立组成、环和腔是非常重要的拓扑特征。具有一定连接关系的结构会产生在内在不变的拓扑特征。持续同调可以记录原子尺寸变化过程中,系统拓扑不变量的开始和结束,通过条形码(barcode)可以进一步将其可视化,也称为拓扑指纹。
北京大学深圳研究生院新材料学院潘锋课题组与美国密歇根州立大学数学系魏国卫教授课题组合作首次将以持续同调为基础的数学方法引入材料科学中,利用持久同源将高维空间中的材料结构映射到低维拓扑空间,从而更方便地研究结构与性能之间的关系。团队将持续同调方法引入碳硼烷体系的分析,通过对这些结构的拓扑指纹的分析,建立起基于拓扑不变量及其持久长度的模型。利用拓扑不变量(β0,β1和β2)定量分析和预测了碳硼烷结构以及其对应BnHn2-的相对能量。通过利用拓扑不变量(β0,β1和β2)的平局长度特征,可以将该拓扑信息与BnHn2- (n = 5~20)的相对能量进行精确拟合,Pearson相关系数为0.977,而相应的碳硼烷结构C2Bn - 2Hn (n = 5~20)的Pearson相关系数为0.937,如图1。该工作以令人满意的精度证明了持续同调方法应用于多原子体系的的可行性,提供了一种新型的团簇结构描述符(Chinese J. Struct. Chem. 2020, 39(6), 999-1008)。在未来的工作中,我们可以利用更有效的机器学习方法,根据持续同源得到的拓扑信息,建立结构与性质之间的关系,实现更大原子数量的碳硼烷结构的预测。
同时,团队在锂团簇能量预测方面,除了使用拓扑不变量来提取团簇结构的拓扑几何信息,对于原子间的短程作用和长程作用等数据信息,还进一步提出了持续独立原子对(PPI)来计算“生长”过程中每对原子(或点)的独立性。最初,所有的原子都是不相连的,PPI条的数目等于独立原子对的数目。随着生长ϵ参数的变大,一些原子对连接起来,它们的PPI合并。我们提出的PPI条码比β0更具信息量。如图2所示,它可以与拓扑不变量一起用于描述材料结构。随后基于提取的拓扑特征,构建团簇能量预测机器学习模型。最终,仅仅利用小型团簇结构构成的数据集来训练所得的模型即可对中型和大型团簇形成非常高的预测精度。该模型可用于团簇结构的快速筛选,加快最稳定团簇结构搜索的速度。相关成果发表在隶属Nature Index的知名杂志The journal of physical chemistry letters(2020, 11, 4392)上。
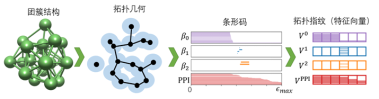
图2基于持续同调与持续独立原子对的锂团簇能量预测
在无机材料的能量预测方面,由于持续同调仅提供全局的拓扑结构信息,对于无机化合物包含大量不同元素和不同结构的体系没法直接使用。因此,团队提出了原子特殊的持续同调(ASPH),它考虑的是晶胞中每个原子周围不同环境构成的持续同调,在拓扑不变量中嵌入原子信息。基于ASPH方法表达的结晶化合物拓扑特征可以构建晶体能量预测模型,使用拓扑表达的结构特征配合元素特征可以实现MAE仅为61meV/atom的结果(图3)。此外基于预测结果,团队还对预测偏差较大的结构进行了详细的讨论与分析,发现了异常通常存在于特殊的氧化态与畸变的结构之中,增加了此类异常的认识与理解。相关成果最近发表在Nature 集团旗下的知名杂志npj Computational Materials(2021,7, 1-8)上。

图3结晶化合物拓扑特征可以构建晶体能量预测模型
拓扑数学是一个强大的工具,可以通过变化的尺度定性分析数据结构,借助机器学习方法,便可以构造有高精度的材料预测机器学习模型用于团簇结构分析、团簇结构搜索以及晶体结构能量预测等诸多材料结构规律的科学研究中,有助于加速材料的发现与应用。
该些工作是在潘锋教授和魏国卫教授的共同指导下,第一作者分别是北京大学深圳研究生院新材料学院研究生陈冬、陈鑫和江毅,他们与团队成员协作共同完成研究,该工作得到了国家材料基因组重点研发计划和深圳市科技计划项目的大力支持。


