利用太阳能驱动光催化无氧脱氢反应,可在100%原子利用率下同步制备高附加值有机化学品与氢气,是实现可持续能源转化与绿色化工合成的核心策略。现有单原子催化剂虽在C(sp3)–H键选择性活化中表现出优异活性,但其活性位点呈空间孤立且均一分布的特征,往往引发电荷分离效率低下、质子还原与C–H 键氧化的协同作用匮乏及反应动力学失衡等问题,难以同时独立调控耦合光催化循环中两个半反应的吸附能与反应路径。双原子催化剂(尤其是异核双原子对)作为单原子催化剂的进阶体系,可依托原子间协同效应实现更精准的反应调控;然而,现有协同机制(如电子结构调控或功能分工模型)仍局限于单一反应路径的优化,尚未在原子尺度上实现氧化还原半反应的完全解耦与空间-功能双分离。

本研究以构建原子尺度空间与功能完全分离的双活性位点为核心出发点,设计了一种锚定在CdS上的PtAg DAC,用于驱动甲苯的光催化无氧转化反应,具体亮点如下:1)创新催化剂结构设计:摒弃单原子催化剂的孤立位点模式与传统双原子催化剂的单向调控机制,构建 PtAg–S 桥连催化单元,形成内建电场,实现电子在Pt位点富集、空穴在S位点局域化的不对称电荷分布,为氧化还原半反应的解耦提供结构基础。2)提出 “双位点平行反应路径” 新机制:基于PtAg DAC的独特结构,实现活性位点的精准功能分工 —— 富电子Pt位点专一催化质子还原制氢,缺电子Ag位点则调控相邻富空穴 S 位点,协同介导甲苯C (sp³)–H键活化与氧化偶联生成1,2 -二苯乙烷;该机制将传统单一速率决定步骤拆分为两个低能垒平行反应,大幅降低整体反应能垒(从3.02 eV降至 1.31 eV)。3)实现催化性能的显著突破:PtAg DAC的1,2 - 二苯乙烷生成速率达32.8 μmol g-1 h-1,选择性为77.35%,性能优于单金属催化剂达3.1倍;同时产氢速率达 89.8 μmol g-1 h-1,分别是Pt、Ag单原子催化剂的2.1倍和6.5倍。4)提供普适性催化剂设计原则:本研究不仅从原子尺度揭示了双原子协同作用的本质机制,更为复杂氧化还原耦合反应中高效光催化剂的设计提供了全新的理论指导。
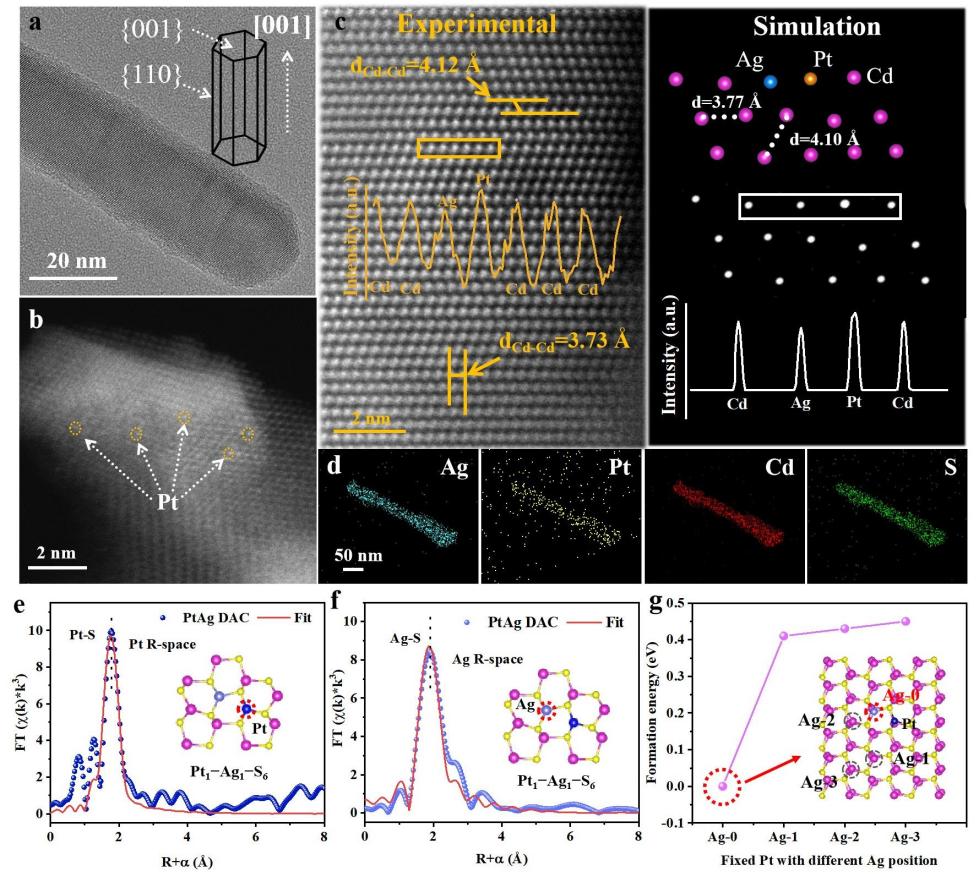
PtAg双原子催化剂的结构表征
要点:本研究通过原位热浸渍法在CdS纳米棒上合成了负载量可控的PtAg DAC。结构表征表明,CdS晶相保持不变,且未观察到金属颗粒或团簇,证实了Pt和Ag的原子级分散。HAADF-STEM图像显示亮点成对分布,原子间距分析揭示了~3.35 Å的Pt-Ag短程相互作用,直接证明了Pt-Ag配对结构的形成。EXAFS分析进一步确认了Pt-S和Ag-S配位环境,配位数约为3,且无金属-金属键,XANES模拟结合EXAFS定量拟合,确认了Pt1-Ag1-S6的结构模型。理论计算表明该双原子构型具有负的形成能和良好的热力学稳定性。
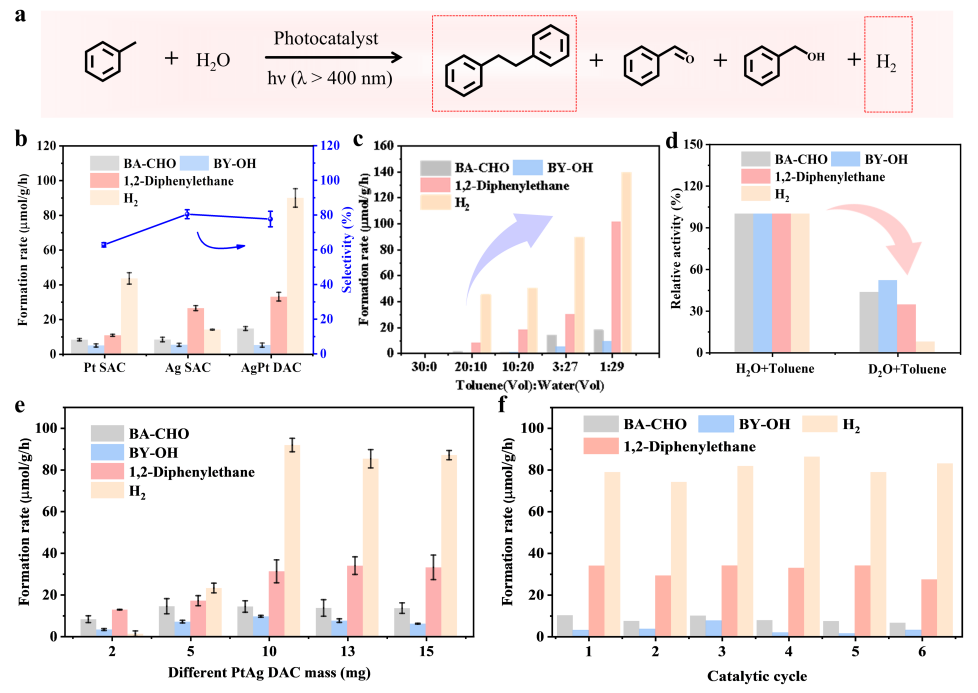
光催化性能及稳定性评估
要点:1). 双原子协同效应与性能优势,通过系统比较,证实PtAgDAC在耦合反应中具有显著协同效应。其产氢速率(89.8 μmol g-1 h-1)和1,2-二苯乙烷生成速率(32.8 μmol g-1 h-1)均远超对应的单原子催化剂,表明双位点分别主导了不同的半反应:富电子的Pt位点专司质子还原产氢,而Ag位点则促进了甲苯的氧化脱氢偶联反应。2). 水溶剂的关键作用与反应机理验证,水在反应中扮演了不可或缺的角色。提高水含量能同时促进两个产物的生成。关键的同位素实验(D2O)证明,生成的氢气中的氢原子部分来源于水溶剂,而非甲苯,这直接证实水是产氢反应的质子源,并参与了C–H键的活化过程。3). 催化剂的优异稳定性与结构保持,PtAg DAC在六次循环测试中表现出优异的稳定性,产物产率无显著下降。这种稳定性归因于双原子位点作为高效的电荷提取中心,能快速转移光生空穴和电子,从而抑制了载流子复合和催化剂的光腐蚀。
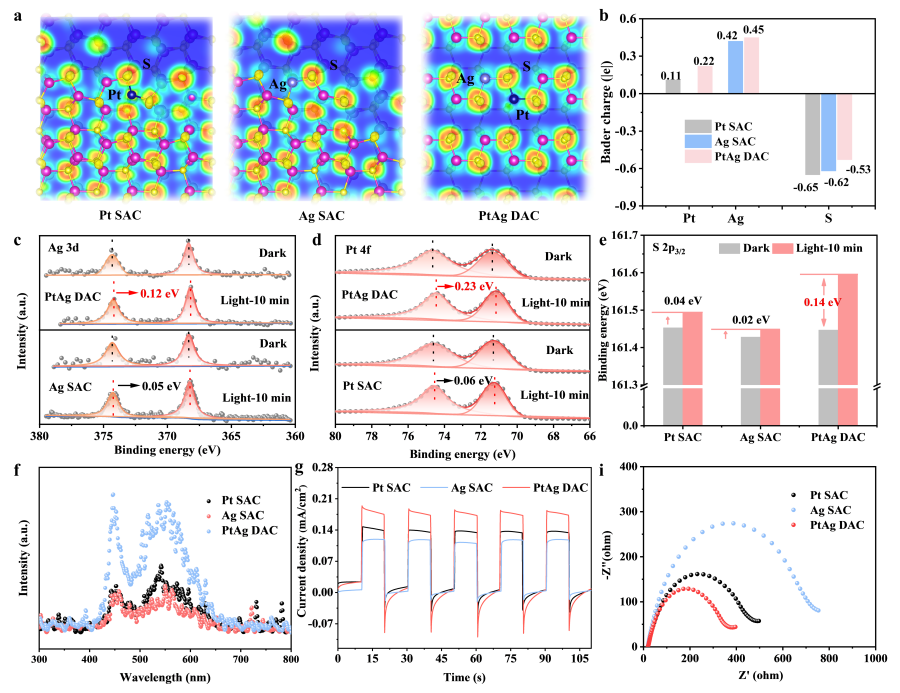
PtAg 双原子催化剂中的电子结构调制及电荷分离动力学
要点:1). PtAg-S桥单元引发独特的电子再分布与内部电场,DFT计算表明,PtAg双原子对的形成导致显著的电荷重排。电子主要从Ag向Pt-S界面转移,使Pt正电荷显著增加(从+0.11 |e|增至+0.22 |e|),远高于Ag的微小变化。这一过程在Pt-Ag-S桥单元内产生了一个由Ag指向Pt的内部电场。2). 内部电场驱动电荷的定向分离与功能分工,该内部电场成为光生电荷分离的强力驱动器。它持续将光生电子导向Pt位点,使其成为富电子的质子还原(产氢)中心;同时将光生空穴导向Ag及其相邻的S原子,使Ag-S中心成为缺电子的C(sp³)–H键活化(氧化)位点。原位XPS数据在光照下观测到Ag、Pt结合能下降(得电子)而S结合能上升(失电子),直接验证了这种电荷的定向积累与消耗。3). 光谱与电化学分析证实了增强的电荷分离与迁移动力学,稳态荧光光谱显示PtAg DAC的发光强度最弱,表明其辐射复合受到抑制。时间分辨荧光寿命测试表明PtAg DAC具有更短的寿命,意味着更高效的电荷转移和提取。此外,电化学阻抗谱显示其电荷转移电阻更低,光电流响应更强。这些结果共同证实,Pt-Ag双原子位点不仅促进了定向电荷分离,还显著加速了光生载流子向各自活性位点的迁移,从而整体提升了光催化氧化还原效率。
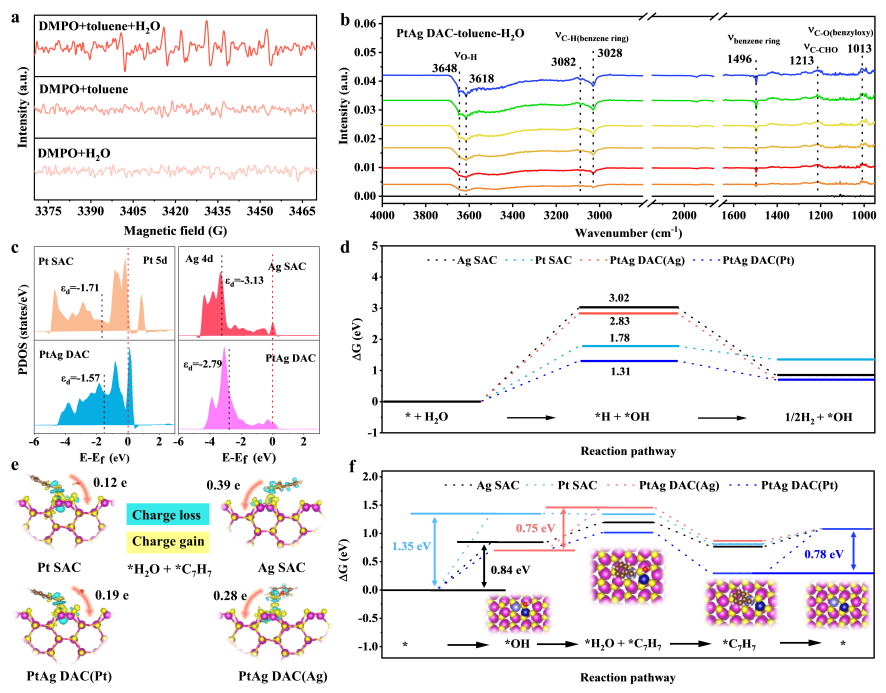
PtAg 双原子催化剂表面反应路径的机理研究
要点:1). 光谱表征明确了反应路径,电子自旋共振(ESR)测试捕获到苄基自由基(C7H7·)信号,却未检测到羟基自由基(·OH)的存在;结合自由基清除实验结果,证实甲苯的活化遵循直接空穴氧化路径,而非羟基自由基介导的氧化机制。原位红外光谱(in situ FT-IR)的表征结果进一步佐证了上述路径:光照下甲苯特征峰消失但未直接检测到1,2-二苯乙烷的中间信号,表明目标偶联反应由苄基自由基直接发生耦合反应完成。2). 理论计算阐明了水分子的关键调控作用及双原子位点的协同催化机制,Pt和Ag的d带中心在双原子体系中均上移,这一现象源于 Pt 与 Ag 之间的强电子耦合作用,最终促使 Ag 位点呈现缺电子态(有利于 C-H 键的氧化活化)、Pt 位点呈现富电子态(有利于H2的析出反应)。反观单原子催化剂(Pt SAC 与 Ag SAC),其催化效率受限于*OH转化的高能垒步骤(Pt SAC: 1.35 eV; Ag SAC: 0.84 eV)。而PtAg DAC通过双原子位点协同,将水辅助的甲苯氧化过程拆分为两个能垒更低并行反应步骤:其一,在Ag-S(桥位)点进行*OH → *H2O + *C7H7(能垒0.75 eV),在Pt-S桥位点进行*C7H7脱附(能垒0.78 eV)。这种反应路径的重构从根本上提升了催化反应的动力学速率。3). 电子结构根源,Ag位点能够调控S(桥位)位点的氧化能力,并通过化学键将这种空穴富集特性高效传递给相邻的S(桥位)原子,从而显著增强S(桥位)原子对C(sp3)-H键的活化能力。而 Pt 位点则维持较短的键长与强共价吸附特性,主要充当电子富集中心并专一性地介导产氢反应,并非高效的氧化活性中心。
总结与展望:
针对光催化芳烃甲基无氧活化及C-C偶联反应中氧化还原位点协同效应匮乏的关键瓶颈,本研究创新性地设计了一种锚定于硫化镉(CdS)载体的铂-银双原子催化剂(PtAg DAC)。该催化剂借助 PtAg-S桥连结构构建的内建电场,在原子尺度上实现了氧化位点(Ag-S)与还原位点(Pt)的空间-功能双重解耦,成功将水辅助甲苯转化-产氢耦合反应的能垒从3.02 eV显著降至1.31 eV,最终实现了1,2 -二苯乙烷(32.8 μmol g-1 h-1)与氢气(89.8 μmol g-1 h-1)的高效联产。该催化剂的催化性能显著优于单原子对照样品,且具备优异的结构与催化稳定性。本工作不仅阐明了双原子催化剂“空间-功能解耦”的协同催化新机制,更为复杂耦合光催化反应高效催化剂的理性设计提供了全新范式;未来研究可进一步拓展双金属组合类型、借助超快光谱技术解析电荷转移动力学机制,并将该催化策略拓展至更多惰性C-H键活化的复杂反应体系中。
论文链接:https://advanced.onlinelibrary.wiley.com/doi/10.1002/adma.202521903
以上研究工作得到国家自然科学委面上项目(No. 22579005)、海外高层次人才项目、北京大学深圳研究生院启动基金的资助,同时得到了曙光超级计算中心和上海同步辐射光源的支持。访问学者唐永华博士为论文第一作者,北京大学深圳研究生院周鹏助理教授与湘潭大学李红星教授为论文通讯作者。


